What is Holoprosencephaly?
Holoprosencephaly, often referred to as HPE, is a disorder that occurs when the front part of the brain, known as the forebrain, fails to divide properly down the middle. This condition can lead to a variety of issues, both inside the head and on the face. These issues can range from problems with brain function to abnormalities in facial features. It’s the most common disorder related to the development of the forebrain. Sometimes, HPE occurs randomly without any clear cause, while other times, it’s linked to a specific genetic syndrome. This issue typically forms when the baby is still an embryo, roughly 3 to 4 weeks after conception.
The HPE condition has been categorized into 3 different types based on how severe it is.
First, there’s “Alobar”, which is the most severe form and the most frequent one. In this category, the forebrain does not split at all, resulting in a single central chamber in the brain instead of two separate sides.
Second, there’s “Semilobar”, in which the forebrain partly divides.
Third, there’s “Lobar”, where the forebrain nearly fully divides.
Besides these three, there are two rare variants. One, referred to as “Middle interhemispheric variant” or “syntelencephaly”, where only the back parts of the frontal and parietal lobes – regions of the brain – join abnormally along the midline. Another one is the “Septo-preoptic variant”, which is the mildest form, and the fusion along the midline is limited to small areas in the front of the brain.
What Causes Holoprosencephaly?
Holoprosencephaly (HPE), is mostly studied in individuals with regular genetic composition but with unusual chromosome patterns. These cases rarely happen alongside other birth defects. In majority of babies born with HPE, it’s typically not linked to genetic conditions or chromosome issues. Instead, it might be due to environmental causes, genetic deletions, changes in genes associated with HPE or other unknown factors.
Understanding HPE involves looking at a few categories:
When it comes to genetic causes, there’s one main group that doesn’t involve a syndrome: the most common example of this is anomalies of the SHH gene in patients with HPE who also have defects in the middle line of their bodies. Other important genetic causes include changes in the ZIC2, SIX3, and TGIF genes. Here’s why these genes are important:
* The ZIC2 gene – helps organize a structure known as the anterior notochord in early development.
* The SIX3 gene – is involved in organizing the front-to-back positioning of structures in the brain.
* The SHH gene – helps shape the forebrain and the eye’s optic cup from the front-middle area of the neural tube.
* Bone morphogenetic protein – is involved in the shape of the skull and face.
* Fibroblast growth factor – also determines the shape of the skull and face.
* The TGIF genes – help regulate how SHH functions.
Syndromic causes of HPE can account for 32% to 42% of cases. These typically happen because the number of chromosomes is anomalous, most often resulting in tripling of the 13th chromosome (Patau syndrome), the 18th or in triploidy. These lead to various birth defect syndromes, like Steinfield syndrome, Kallman syndrome, Hartsfield syndrome, and others.
Certain genes can also contribute to the development of HPE. Some, called driver genes, are critical to how the condition develops while others can modify or predispose to HPE.
Non-genetic causes can be related to various maternal conditions, such as diabetes before pregnancy. It has been observed that regular folic acid supplements for pregnant mothers could have protective effects against HPE. Animal studies have also pointed towards potential harmful influences leading to HPE, such as specific environmental factors or exposure to certain substances like alcohol, retinoic acid, fungus-produced toxins, certain drugs, and others. Environmental factors including certain substances found in smoked cigarettes, charred meats, cannabis, and some pesticides often found in household dust have also been linked to the occurrence of HPE.
Risk Factors and Frequency for Holoprosencephaly
Holoprosencephaly (HPE) is a condition that occurs commonly in about 1 in 250 developing embryos. However, the number of cases seen in newborns ranges from 1 in 8,000 to 1 in 16,000 live births globally. The number of cases remains the same across different global populations.
- In the United States, some groups, such as African-American, Hispanic, and Pakistani communities, slightly more often have this condition.
- The likely reason for this increased rate is a lower frequency of prenatal diagnosis and related pregnancy terminations within these specific groups.
Signs and Symptoms of Holoprosencephaly
Holoprosencephaly (HPE) is a disorder connected to abnormal genes that play a role in the development of the head and face. Symptoms of HPE usually vary from mild to severe. A patient with mild symptoms might only have small facial defects such as a single upper central tooth, but with normal brain function. However, severe cases may involve major defects in the face and brain, such as cyclopia (having one eye), or proboscis (having a nose that looks like a tube and has only one nostril).
The more severe the facial symptoms, the more serious the damage to the brain is likely to be. These symptoms are usually associated with the alobar form of HPE, and they include:
- Significant microcephaly (an unusually small head)
- Cyclopia: One single eye in the middle of the face, or no eye at all
- Proboscis: A tube-like nose with one nostril, which can be present or not
- Cebocephaly: A combination of closely-set eyes and a nose with one nostril
- Ethmocephaly: A proboscis and closely-set eyes
On the other hand, less severe facial symptoms are usually related to the lobar form of HPE and include:
- Hypotelorism: Eyes that are too close together
- Midface hypoplasia: Underdeveloped middle facial features
- A flat nasal bridge
- Cleft lip and/or palate
- A missing nasal columella
Note, though, that severe facial defects such as ocular colobomas (gaps in parts of the eyes) could be associated with the alobar form, but when they exist on their own, it’s usually a sign of the lobar/middle interhemispheric form of HPE. The single maxillary central incisor-microform variant is also considered a milder form of the disease. Lastly, individuals with a mild form of HPE usually have a family member with a more obvious case of HPE and typically exhibit only facial abnormalities without accompanying neurological issues.
Testing for Holoprosencephaly
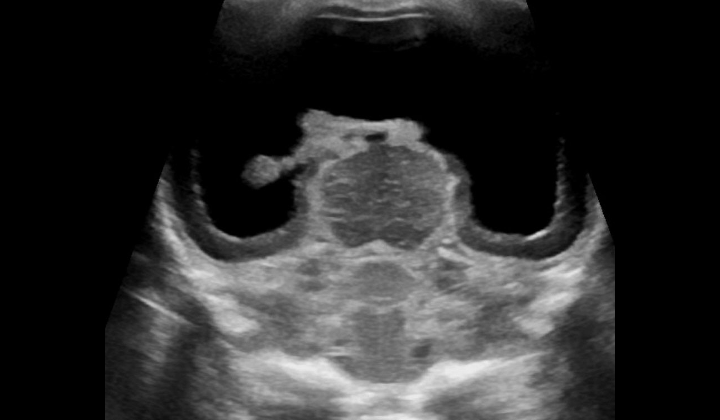
If a child is showing signs of unusual development, has a family history of certain conditions, and has been in certain environmental factors, they may need to be examined for what’s known as an intracranial anomaly, essentially an abnormality in the brain. This examination could actually start before they are born. As early as the first trimester of pregnancy, a method called non-invasive prenatal testing (NIPT) can be used to identify the likelihood of a condition known as fetal trisomy 13. The examination process includes using imaging methods like ultrasound and MRI.
For severe cases of a condition called HPE, an ultrasound can spot the abnormality as early as the first trimester, while an MRI is often used for diagnosis in the third trimester. Once the baby is born, an ultrasound is often used first if the soft spot on their head, called the anterior fontanelle, is still open. As the child gets older, a CT scan or MRI may be considered. However, these tests come with risks, such as exposure to radiation from the CT scan or the risk of excess sedation with MRI.
Various patterns can be observed during the imaging tests, which represent different forms of HPE. For example, in an alobar HPE, there are a number of signs including missing parts of the brain structures and a fused deep gray nuclei (part of the brain), In the semilobar form, the frontal lobes of the brain aren’t separated and certain parts of the brain are absent or fused. Similar patterns and missing or fused brain structures are observed in the lobar form, middle interhemispheric variant (syntelencephaly), and microform HPE.
There are also certain features or “signs” that doctors look for in the images. These include a “horse-shoe shaped mono ventricle” on the images, the absence of a feature known as the butterfly sign, and a missing normal cavum septi pellucidi, which should normally be present in certain types of HPE. In addition, the visualization of what’s referred to as a “Snake under the skull” sign can suggest the presence of lobar HPE.
Electroencephalography, which records electrical activity of the brain, can also find certain patterns in people with HPE. These include multifocal spikes that often become a pattern called hypoarrhythmia, and a steady high-voltage alpha-theta monorhythmic activity when the child is awake in the newborn stage, which changes when the child sleeps.
Once HPE is confirmed, doctors would check if there are any related conditions, also known as a syndromic association. If one is found, management may include evaluating any coexisting issues that might need additional medical or surgical attention. Genetic testing may also be carried out to see if any chromosomal or genetic anomalies associated with HPE are present. If they are, the child’s parents could be offered genetic counseling for future pregnancies.
Treatment Options for Holoprosencephaly
Managing Holoprosencephaly (HPE), a complex brain condition, requires a team approach. Treatment focuses on relieving symptoms and managing complications affecting different organ systems:
* Seizures: Roughly half of all children with HPE experience at least one seizure. In some cases, a combination of anti-seizure medications may be needed to manage these seizures.
* Enlarged Head with Fluid on the Brain: Sometimes, children with HPE have an enlarged head due to excess cerebrospinal fluid, a condition known as hydrocephalus. If this occurs, a surgical procedure called shunt surgery can be done to drain the excess fluid and relieve pressure on the brain.
* Movement Disorders: Some children with HPE have motor anomalies such as stiffness (spasticity) or challenging movements (dystonia). Medicine can help manage these symptoms, along with physical and occupational therapy.
* Difficulty Eating and Breathing: Structural issues like cleft lip or cleft palate can make it difficult for children to eat and breathe, leading to aspiration pneumonia. These structural issues may require surgical correction.
* Hormone Disorders: Given that HPE impacts the development of the hypothalamus, a part of the brain that helps regulate hormones, some children may have hormone deficiencies. The most common is diabetes insipidus, which involves extreme thirst and heavy urination, but other deficiencies can also occur. Supplements, such as prednisone, thyroxine, and growth hormone, might be given to manage these deficiencies.
* Digestive Problems: Some children with HPE have slow gastric and intestinal motility and gastroesophageal reflux due to poor nerve migration to the gastrointestinal tract. In severe cases, a gastrostomy tube may be needed to help feed the child. Medications and procedures for reflux may be needed in other cases.
What else can Holoprosencephaly be?
When trying to diagnose a medical condition known as HPE, doctors have to keep in mind other medical conditions that could potentially show similar symptoms. These include:
- Septo-Optic dysplasia: This condition comes with symptoms like enlarged ventricles, failure of the hypothalamic-pituitary axis, degeneration of the optic nerve, and an absent septum pellucidum.
- DiGeorge syndrome: The abnormalities associated with this syndrome include a cleft palate and hypertelorism. It can also be associated with cardiopulmonary anomalies like aortic arch and conotruncal defects. Other symptoms include T-cell deficiency and hypocalcemia resulting from parathyroid hypoplasia.
- Hydranencephaly: By conducting a CT scan of the head, this condition shows the absence of bilateral cerebral hemispheres with no cortical mantle, and a fluid-filled cavity. Since the falx is still intact, HPE can be ruled out.
- Porencephalic cyst: In MRI scans, this condition shows up as an abnormal accumulation of CSF within the brain parenchyma. In this case, the cerebral lobes are not absent/midline fused like in HPE.
- Arachnoid cyst: This condition occurs alongside HPE or could be the only condition present. These cysts usually occur as a result of trauma or could be inherited in an autosomal manner. Unless HPE is also present, facial defects are not usually observed in this condition.
What to expect with Holoprosencephaly
In moderate to severe cases of HPE (Holoprosencephaly, a brain development disorder), the prognosis is often not good. The mortality rate is high, with a third of newborns typically passing away within the first 24 hours after birth. By the first month, this increases to 58%. Between the fourth and fifth months, half of the babies with this condition unfortunately do not survive, and by their first year, this increases to 70-80%. Only a small number of these newborns manage to survive until adulthood.
A recent study from a large population registry surprisingly showed a slightly more optimistic picture, with a 1-year survival rate at 58.1%, and a 10-year survival rate at 36.9% for children with HPE.
For those with mild to moderate cases of HPE, they usually survive until adulthood, but they often live with complications. Interestingly, about half of people with a specific type of HPE, known as lobar HPE, have the ability to walk and communicate verbally.
Possible Complications When Diagnosed with Holoprosencephaly
People with HPE (Holoprosencephaly) can experience neurological issues, such as epilepsy, movement disorders characterized by low muscle tone or abnormal muscle contractions, irrespective of any visible seizure activity. Hydrocephalus, a condition where excess fluid builds up in the brain, is often seen in an HPE type called ‘alobar variant’. This can lead to an abnormally large head, unlike the small head size typically suspected in HPE.
HPE can also cause issues in the digestive system, such as slow stomach and colon movements and acid reflux, due to problems in nerve development.
In people with HPE, the brain structure responsible for regulating vital functions and hormones (the hypothalamus) may not form correctly. This can lead to various hormone imbalances due to deficiency in pituitary gland hormones and abnormalities in vital functions like thirst, hunger, and body temperature. Problems linked to the back part of the pituitary gland, like diabetes insipidus, are more common and can be identified through regular screening for electrolyte levels. Hormone deficiencies linked to the front part of the pituitary gland may lead to conditions like underactive thyroid (hypothyroidism) and inadequate cortisol (a steroid hormone) production (hypocortisolism), which can be deadly. Lack of growth hormones and reproductive hormones can result in stunted growth and delayed sexual maturity, respectively.
Typical HPE Complications:
- Epilepsy
- Motor impairment
- Hydrocephalus leading to larger head size
- Poor gastric and colonic movement
- Gastroesophageal reflux (acid reflux)
- Under formation of hypothalamus leading to vital function abnormalities
- Pituitary hormone deficiency leading to potential hormone imbalances
- Diabetes insipidus
- Hypothyroidism and hypocortisolism
- Stunted growth and sexual immaturity
Preventing Holoprosencephaly
If your child is showing symptoms or milder signs that could suggest a genetic condition, doctors often recommend genetic testing. This test can tell us if your child has certain changes in their genes. Deciding whether to test can be difficult, though, because some conditions can have a wide range of different symptoms. It’s further complicated when multiple genes are involved or when a problem is caused by several different changes in genes, which can make diagnosis very challenging.
We’ve also seen some instances where a genetic mutation, or change, appears out of nowhere in a child without any similar issues noticed in the parents’ genes. In these scenarios, the odds of having another child with a condition called holoprosencephaly (HPE), where the brain doesn’t divide properly, are much lower in future pregnancies.











