What is Creutzfeldt Jakob Disease (CJD)?
Creutzfeldt-Jakob disease (CJD) is a rare, fast-progressing brain disorder caused by prion proteins, which are infectious agents made up primarily of protein. This disease always results in death and very rarely can be passed from one person to another. It takes a long time before symptoms show up. CJD was first identified almost a hundred years ago, in the early 1920s, by Hans Creutzfeldt and Alfons Jakob. Later, another researcher named Clearance J. Gibbs began to use this term because its abbreviation “CJD” reflected his initials.
CJD primarily affects the central nervous system (CNS), which is made up of the brain and spinal cord. The main working part of the CNS is the neuron, a special type of cell that sends, holds, and receives information. Unlike many other cells in the body, neurons in the CNS cannot naturally regenerate, though some sections of the brain can partially repair themselves as the brain has stem cells – a type of cell that can become many different types of cells. The CNS is unique compared to other systems in the body because:
– It has a self-regulation system for blood flow in the brain.
– It is protected by the skull.
– It has specific dietary needs.
– It doesn’t have a traditional lymphatic system, which is a network of tissues and organs that help rid the body of toxins.
– The fluid around the brain and spinal cord, known as cerebrospinal fluid, continuously circulates.
– It has minimal immune surveillance, or protection.
– It has its own special methods for responding to injury and repairing tissues.
Neurons in the brain are arranged according to the tasks they perform. Some, located in particular regions, control intentional movements, while others control automatic bodily functions. This, along with the fact that each brain cell communicates with specific parts of the body, helps identify the locations of brain injuries.
Neurons vary in size and structure. They connect with each other through axons and dendrites, structures that allow for communication between cells. Another important feature of neurons is Nissl bodies, which help in the making of protein and neurotransmitters, chemicals that neurons use to communicate. They also contain structures called neurofilaments that maintain the cells’ shape and contribute to nerve signaling.
Additionally, the CNS includes glial cells that aid the neurons. These include:
– Astrocytes: Star-like cells that feed neurons, detoxify nerve cells, guard against harmful substances, and contribute to CNS repair and the formation of scar tissue.
– Oligodendrocytes: Cells that create the insulation (myelin) for nerve fibers in the CNS.
– Ependymal cells: Cells that line the ventricles (fluid-filled spaces in the brain), governing the production and movement of cerebrospinal fluid.
– Microglia: Cells that make up the immune system in the CNS.
In CJD, harmful substances can build up inside the neurons, damaging brain cells. This initially results in generic symptoms that can be hard to recognize, but as the disease progresses, it often causes noticeable neurological problems, particularly sudden, ‘shock-like’ muscle jerks (myoclonus).
What Causes Creutzfeldt Jakob Disease (CJD)?
A prion protein, often shortened to PrP, is a normal neuron protein that mostly exists as flexible spirals and random twists. Prions, also known as “proteinaceous infectious particles,” are proteins that can copy themselves without needing any genetic material. These prions are mainly resistant protein aggregates that reproduce by attaching to normal cell versions of PrP proteins, changing flexible spirals into tightly packed, indigestible folds. These particles are responsible for diseases such as Creutzfeldt-Jakob disease (CJD) and other diseases that cause neurodegeneration like mad cow disease, kuru, and scrapie.
The classification of CJD is based on how it’s transmitted. Sporadic CJD, which is the most common kind and accounts for approximately 85% of cases, happens due to misfolding of normal PrP proteins, and there doesn’t seem to be a particular trigger for this. There are different variations of sporadic CJD like sporadic fatal insomnia and variable sensitivity prionopathy. Genetic CJD is the second most common kind and takes up about 10-15% of cases. This results from a genetic mutation passed down through families. Variations of this type include familial CJD, fatal familial insomnia, and Gerstmann-Sträussler-Scheinker syndrome.
Infectious CJD, which makes up less than 1% of cases, occurs when prions are introduced into the body from an outside source. Subtypes of infectious CJD are kuru, iatrogenic CJD, and variant CJD. Kuru was a disease found amongst the Fore people of Papua New Guinea who used to eat the brains of dead relatives as part of ritualistic cannibalism. This practice was banned in the 1950s. Iatrogenic CJD occurs when prions are accidentally introduced into the body during medical procedures. Variant CJD is linked with eating infected beef, a transmission method similar to mad cow disease. Most recognized cases of variant CJD have been reported in the United Kingdom and France.
Risk Factors and Frequency for Creutzfeldt Jakob Disease (CJD)
Creutzfeldt-Jakob Disease (CJD) is a rare condition affecting about 1 in a million people across the globe every year, with about 350 cases identified annually in the U.S. The most frequent type of this sickness is sporadic CJD. It usually arises around the age of 62 but can occur in other age groups as well. Sporadic CJD affects males and females equally, with 1 to 2 new cases identified per million individuals worldwide every year. Unfortunately, it’s a serious condition – nearly 70% of patients pass away within a year of onset, averaging a lifespan of 4 to 8 months after detection. Up to 90% of patients die within a year.
Genetic CJD is the next most ordinary type, usually found in patients with a family history of the disease and specific inherited gene mutations. Acquired types (that come from exposure to the disease) make up less than 1% of cases, typically seen in younger adults around the age of 29.
Signs and Symptoms of Creutzfeldt Jakob Disease (CJD)
Creutzfeldt-Jakob Disease (CJD) is a disease affecting the brain, leading to different symptoms based on the specific subtype of the disease. This disease’s symptoms are dependent on the parts of the central nervous system (CNS) affected, and can include:
- Mental and behavioral symptoms such as memory problems, concentration and judgment difficulties, and mood changes like depression or anxiety
- Sleep disturbances and visual hallucinations
- Involuntary movements including spasms, tremor, and muscular rigidity
- Problems related to coordination, movement, and balance, like the inability to control eye movements (nystagmus) and unsteady, clumsy movements (ataxia)
Less common symptoms include abnormalities in cranial nerves, peripheral nerve involvement, and dysfunction of the inner ear affecting balance and hearing. If any of these unusual symptoms are present, it’s important to consider other possible diseases as well.
In the early stages of the most common type of CJD (sporadic CJD), patients might experience symptoms like dizziness, tiredness, headache, sleep disorders, and changes in vision. As it progresses, patients tend to develop severe memory and cognitive problems, mood swings, and issues with coordination and movement, which may become disabling. Over time, the condition worsens, leading to a coma-like state and potential death from complications like pneumonia. Most patients are aged between 55 and 75 and typically pass away within months of symptom onset.
- The most common subtypes of sporadic CJD are MM1 and MV1, representing about 70% of cases. These people have symptoms of rapidly worsening dementia and early physical coordination problems.
- The MV2 variant affects about 10% of sporadic CJD cases, typically showing progressive dementia with a strong psychological aspect and lasting about 17 months.
- The VV2 variant often causes issues with physical coordination and lasts around 7 to 9 months.
- The MM2 variant can either impact the thalamus in the brain (“thalamic”), causing rapid physical movements, balance problems, sleep disorders, and cognitive problems; or affect the brain’s outer layer (“cortical”), mainly causing dementia. Both variants typically last around 15 months.
- The VV1 variant, affecting about 1% of cases, usually starts at a younger age and mostly involves dementia. This variant typically lasts just over a year.
Genetic or inherited CJD also exists, often showing up in younger patients than the sporadic type. Initial symptoms often involve changes in behavior and cognition, later followed by physical coordination and movement problems. Just like sporadic CJD, inherited CJD can also be fatal. One specific subtype, Gerstmann–Straussler–Scheinker syndrome, can progress slowly, with death potentially delayed by up to 10 years.
Finally, patients with variant CJD are usually younger than those with sporadic CJD. They often present with psychological symptoms, mood changes, and abnormal sensations of pain (called dysesthesias) initially. Movement disorders can develop early on, but severe cognitive decline like dementia tends to be a late sign.
Testing for Creutzfeldt Jakob Disease (CJD)
Creutzfeldt-Jakob disease can be difficult to diagnose because its symptoms are similar to those of other conditions that cause rapid cognitive decline. Your doctor might order a series of blood and urine tests to help determine the cause. These can include tests for conditions like Lyme disease, HIV, and other autoimmune diseases, as well as measures of inflammation and general measures of your body’s health, like a complete blood count and urinalysis.
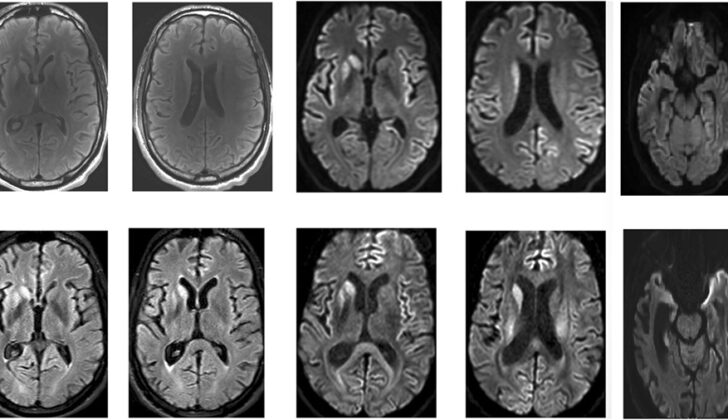
But blood and urine tests alone might not be enough. Sometimes, the doctor might also need to take a sample of your cerebrospinal fluid, which is the fluid that surrounds your brain and spine. These tests can show whether there are proteins associated with prion diseases like Creutzfeldt-Jakob disease. Also, doctors might use imaging techniques, like computed tomography (CT) scans or magnetic resonance imaging (MRI), to get a clear picture of your brain.
An MRI can give considerable details about certain areas of the brain affected by Creutzfeldt-Jakob disease. The frequency of brain imaging in combination with other tests such as laboratory tests and electroencephalogram (EEG) can increase the accuracy of the diagnosis.
However, not all the tests are specific to Creutzfeldt-Jakob disease, so it can be hard to make a definitive diagnosis. Some proteins tested, including the 14-3-3 protein, are often found in the cerebrospinal fluid of patients with Creutzfeldt-Jakob disease, but they are not specific to the disease.
A relatively new diagnostic test introduced in 2015 called “second-generation Real-Time-Quaking-Induced Conversion (RT-QuIC)” is more sensitive and specific for detecting Creutzfeldt-Jakob disease. This test accurately recognises the abnormal proteins in the cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. However, the guidelines for diagnosis of Creutzfeldt-Jakob disease have not yet included this new diagnostic tool.
Brain biopsy or post-mortem brain examination is the gold standard for confirming Creutzfeldt-Jakob disease diagnosis. However, due to the risks involved in brain biopsy and the fact that confirming the diagnosis doesn’t change the potential outcome for the patient, it is performed only when a reversible condition is suspected.
It’s also recommended to be cautious while handling body fluids and tissues from patients with Creutzfeldt-Jakob disease until it’s confirmed the disease isn’t present.
Treatment Options for Creutzfeldt Jakob Disease (CJD)
CJD, or Creutzfeldt-Jakob Disease, doesn’t have a definitive cure at this time, meaning treatment mainly involves providing comfort and support to those affected. While many drugs have been tested for the treatment of CJD, none have shown significant results so far. That said, in laboratory experiments on rodents, a substance known as intraventricular pentosan polysulfate seems to interfere with the formation of destructive PrPSc proteins. There were some cases where patients treated with this substance lived considerably longer, from 37 to 114 months. Nonetheless, we still need more research to find a true cure for this deadly condition.
Early identification of a mutation in the PRNP gene can be beneficial for families who are at risk for the genetic form of CJD. This early detection can help those with the condition to plan for end-of-life circumstances sooner. Providing emotional support and care that improves comfort can enhance the quality of life for these patients. Furthermore, genetic counseling and thoughtful family planning can help prevent passing the PRNP mutation and, subsequently, the disease, to future generations.
What else can Creutzfeldt Jakob Disease (CJD) be?
Rapidly progressive dementia (RPD) can be caused by a wide range of conditions. These include problems with blood vessels, degenerative brain diseases, immune system disorders, infections, blood clots, cancer, treatment side-effects, and harmful substance-related conditions.
For example, stroke, multiple small strokes, or conditions like cerebral myeloid angioplasty and hypertensive encephalopathy, which affect the blood vessels in the brain, can lead to RPD. Inflammation of the blood vessels (vasculitis) and a type of lymphoma that affects the blood vessels can also result in RPD.
Doctors can usually tell these conditions apart from Creutzfeldt-Jakob Disease (CJD), another cause of RPD, by performing a detailed medical examination. This includes imaging studies, such as CT or MRI scans, which can help rule out problems related to blood vessels and cancer. Blood tests and examining the cerebrospinal fluid (the fluid that surrounds the brain and spinal cord) can help identify immune, infectious, metabolic, and degenerative brain conditions.
What to expect with Creutzfeldt Jakob Disease (CJD)
The overall outlook for CJD, also known as Creutzfeldt-Jakob Disease, is unfortunately very poor, even with the advances in understanding this disease. The condition is always fatal. Typically, death takes place within one year after the symptoms first appear, although there are exceptions in some cases.
Possible Complications When Diagnosed with Creutzfeldt Jakob Disease (CJD)
Creutzfeldt-Jakob disease (CJD) comes with many challenges, both physically and emotionally. People with CJD often start to pull away from their loved ones and over time, they may even lose the ability to recognize or relate to them. As the disease progresses, patients also lose the ability to take care of themselves and in many cases, they may end up in a coma. It’s important to note that CJD is always fatal.
Preventing Creutzfeldt Jakob Disease (CJD)
CJD, also known as Creutzfeldt-Jakob disease, is a deadly condition that worsens over time. Thankfully, it is a rare disease in the United States, so the chances of getting it are extraordinarily small. The main approach is to prevent the disease from spreading. To this end, blood donation centers prevent close family members of individuals diagnosed with CJD from donating blood. Similarly, hunters who might eat or handle elk or deer meat are advised to get the meat tested before they consume it. Families who have a higher risk for the inheritable type of CJD might consider using birth control to prevent passing the disease on to their children.











