What is Fanconi Anemia?
Fanconi anemia is the most common reason for inherited failures in the bone marrow. It’s a rare genetic disorder that affects all three types of blood cells. This disorder normally happens when someone inherits two faulty copies of a certain gene, which can be caused by mutations like defects in the gene, duplicates, wrong splicing, and deletions. These faulty genes interfere with the ability of cells to repair themselves, causing an increase in damaged genetic material in our bodies.
Fanconi anemia proteins typically keep our genetic material healthy and help with cell replication by mending a particular type of DNA damage known as interstrand crosslinks (ICLs). These ICLs keep the DNA strands together and maintain DNA health. However, with Fanconi anemia, cells can’t properly repair DNA damage, resulting in unstable genetic material, a reduced number of all types of blood cells, and an increased vulnerability to harmful substances, UV radiation, deformities, and a higher risk of cancer. Fanconi Anemia can also affect almost all organs in the body.
Often, Fanconi anemia is categorized as an inherited form of aplastic anemia. Research on other related disorders has improved our understanding of how Fanconi anemia affects bone marrow. Its typical symptoms include breathlessness, chest pain, dizziness, and fatigue. Patients might also have a history of excessive bleeding from wounds and unusual skin spots due to low platelet count. The condition often accompanies other birth deformities and is frequently diagnosed in children, around the age of 7. Patients with Fanconi anemia often show abnormal bone structure. Moreover, it can increase the patient’s risk of developing blood and solid tumors.
Fanconi anemia often shows up in blood tests as a decrease in all three types of blood cells – red blood cells, platelets, and white blood cells. If a patient shows signs of this condition with or without characteristic malformations, or if they have a family history of bone marrow issues or certain genetic disorders, they should be tested for Fanconi anemia. The same recommendation applies to those diagnosed with early-onset tumors or seen to have increased sensitivity to standard chemotherapy. A chromosomal fragility test is usually conducted to confirm the diagnosis of Fanconi anemia. Management of Fanconi anemia consists of supportive therapy, transplantation of blood-forming stem cells, and treatment with male hormones.
What Causes Fanconi Anemia?
Fanconi anemia is a disease that can be inherited from your parents. It’s most commonly passed down in what’s called an autosomal recessive manner, meaning you need to inherit a faulty gene from both of your parents. However, rare cases inherited in an X-linked recessive manner, meaning the faulty gene is carried on the X-chromosome, have also been observed.
There’s even an autosomal dominant type of Fanconi anemia, which means you only need to inherit the faulty gene from one parent to have the disease.
Using DNA sequencing, scientists have discovered what they call “pathogenic alleles,” which are variations of genes that cause Fanconi anemia. These can include point mutations, duplications, splicing defects, and deletions.
People with Fanconi anemia have a problem with their double-stranded DNA repair — essentially, their bodies have a harder time fixing DNA damage. More than 23 genes related to Fanconi anemia, known as Fanconi anemia complementation genes (FANC), have been found. All these genes play a role in the DNA repair process.
Most of these genes are autosomal recessive — again, meaning both parents need to pass on the faulty gene — but a few, like FANCB, are X-linked, and one called FANCR (RAD51), is autosomal dominant.
Risk Factors and Frequency for Fanconi Anemia
Fanconi anemia is a very rare blood disorder. On average, it impacts 1 out of every 136,000 newborn babies, but this rate can range from 1 in 100,000 to 250,000 births. Based on European data, the rate of Fanconi anemia is 4 to 7 per million live births. This condition can affect individuals of all races, but is more prevalent in certain populations.
- Fanconi anemia impacts 1 out of 136,000 newborns on average.
- The rate of this disorder can range from 1 in 100,000 to 250,000 births.
- According to data from Europe, the prevalence of Fanconi anemia is 4 to 7 per one million live births.
- All races can be affected by Fanconi anemia.
- The disorder is more common in South Africans, Sub-Saharan Africans, Spanish Gitanos, and US Ashkenazi Jews, with a rate of 1 in 40,000 births and a carrier frequency of 1 case per 100 people among US Ashkenazi Jews.
- The birth rate of Fanconi anemia is approximately 1 per 30,000 live births.
- The disorder slightly affects males more often than females.
Signs and Symptoms of Fanconi Anemia
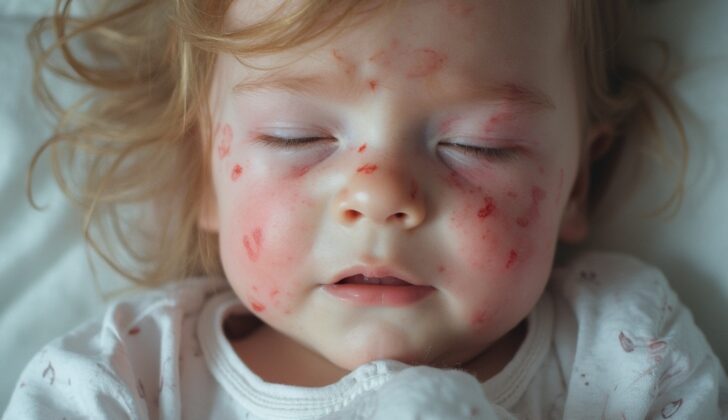
Fanconi anemia is a medical condition that can present with several symptoms. These often include shortness of breath, chest pain, dizziness, and tiredness. People with this condition may also have nosebleeds, petechiae (tiny purple, red, or brown spots on the skin), and bleed excessively from wounds due to low platelet count (thrombocytopenia). Leukopenia – a decrease in white blood cells, can also occur in these patients, making them more susceptible to infections, which can cause fever and flu-like symptoms. Some patients may have had low birth weight, and weight loss might be a sign of underlying cancer. The disease can be more common in populations where marriages between close relatives are prevalent, so family and marriage history should also be considered.
Around 75% of cases of Fanconi anemia are associated with birth defects. A physical examination might reveal several distinctive features. Kids with this condition may be smaller, and over half may have light-brown skin spots known as café au lait spots. Finger and toe abnormalities are common, such as the absence of a thumb, or having an extra toe. Lower limb problems may include club foot, hip dislocation or abnormal bones in the legs. Skull and facial abnormalities are also common, including a smaller head, flat forehead, or an unusually large or small jaw.
- General shortness of breath
- Chest pain
- Dizziness
- Fatigue
- Nosebleeds
- Small, purple, red or brown skin spots (petechiae)
- Excessive bleeding from wounds
- Higher risk of infections
- Low birth weight
- Possible weight loss
Children with Fanconi anemia may also show signs of underdevelopment of the genital areas. Pregnancies can also be complicated by this condition. There might be abnormalities in the eyes, such as drooping eyelids, cataracts, or even blindness. Other features might include abnormalities of the digestive system, heart defects, and signs of pancytopenia – an abnormally low number of all blood cells. These signs can include paleness, cold hands and feet, and easy bruising among others.
Testing for Fanconi Anemia
Fanconi anemia is a disease that causes all types of blood cells to decrease, a condition known as pancytopenia. This is due to a failure in the bone marrow, the place where these cells are produced. Symptoms of this disease typically become noticeable around the age of 7, but increased awareness and improved prenatal screening methods have led to earlier diagnoses. Ensuring early detection is important because it can prevent severe complications.
This disease should be suspected in patients showing symptoms of reduced blood cells or unusual physical defects, and also if there are cases of bone marrow failure in their family history. Moreover, patients who develop tumors early in life or show excessive side effects after regular chemotherapy should also be evaluated for Fanconi anemia. The disease confirmation usually involves a chromosomal fragility test.
During laboratory tests, a complete blood count is performed to determine the levels of red blood cells, white blood cells, and platelets. A larger than normal size of red blood cells or a high fetal hemoglobin level suggests the presence of the disease. Serum erythropoietin, a hormone that promotes the formation of red blood cells, is also typically increased due to the decreased blood cell levels and the poor response of the originating stem cells in the bone marrow. Additionally, bone marrow aspiration and biopsy, a process where a small amount of bone marrow is removed for testing, usually shows lower than normal cell levels and the absence of certain types of stem cells.
In severe cases of pancytopenia, a special diagnostic test known as a chromosomal breakage test is used. In this process, chemicals that cause DNA to break are used to trigger DNA damage in the absence of a repair system. Cultured skin cells also show chromosome fragility and are recommended in patients with negative results of the same test in T-lymphocyte cells, a type of white blood cells or in those who have already undergone bone marrow transplant.
Other tests include cell cycle analysis, a method of identifying cells with impaired DNA repair and a sequencing of the Fanconi anemia gene. Prenatal testing can also be done through amniotic fluid cell analysis or biopsy of the chorionic villus, a part of the placenta.
In addition, doctors may call for imaging tests to identify structural and congenital abnormalities. A skeletal X-ray can help detect bone defects, and an abdominal ultrasound can be used to assess liver and kidney irregularities. Magnetic resonance imaging (MRI) is essential for revealing abnormalities in the brain and pituitary gland.
Treatment Options for Fanconi Anemia
In simpler terms, the main supportive therapy for Fanconi anemia is blood transfusions. Transfusions of red blood cells and platelets can provide immediate relief, but it’s not advisable to have these from family donors due to potential health risks. Patients with low white blood cell count can respond well to certain medicines, but these are given only in severe cases. It’s important to note that heavily relying on transfusions may not lead to good results if the patient had previously undergone a particular cell transplant.
One of the procedures that can effectively treat and prevent certain complications of Fanconi anemia is Hematopoietic Stem Cell Transplantation. This is a process where bone marrow, peripheral blood cells, or umbilical cord blood are transplanted into the patient. It’s best if the transplant comes from a perfectly matched sibling. As per latest studies using this technique, patients suffering from aplastic anemia showed better survival rates. However, the donor’s blood cells need to be tested just to ensure they do not carry Fanconi anemia. Due to several limitations, not everyone has access to this treatment option. It is mostly offered to patients with severe conditions or if other treatment methods fail.
The surge in technological advancements is paving way for alternate approaches to prevent the side effects of transplantation. One such method is using “digital” genomic editing to correct the defective gene. However, these techniques are still under investigation.
Androgen therapy is another option, particularly targeted towards patients who can’t go through stem cell transplantation. Here, certain medicines are used to stimulate the proliferation of the hematopoietic stem cells. While this treatment can’t cure the disease, patients may have a good response, especially the red blood cells. However, the effect on platelets and white blood cells may be less.
Surgery may come into the picture if there are structural deformities that need to be corrected due to Fanconi anemia. For instance, certain issues with the hand should be addressed early in life to avoid hindrance in their daily functioning. Surgeries may be needed to correct birth defects of the heart, rectify issues with the windpipe or the esophagus, and even cure cancers.
Gene therapy is a promising developing treatment that replaces the abnormal gene causing Fanconi anemia with a healthy one. Despite being a prospective cure, it’s not entirely effective and further studies are required in this area.
What else can Fanconi Anemia be?
Fanconi anemia, a blood disorder that leads to bone marrow failure, can look a lot like many other diseases. This makes it really important to rule out other conditions that exhibit similar symptoms.
One such condition is acquired aplastic anemia, which occurs when the bone marrow’s blood-forming stem cells are destroyed – usually by harmful substances. Both conditions share a common symptom in pancytopenia, a condition characterized by reduction of all three types of blood cells. However, unlike Fanconi anemia, acquired aplastic anemia doesn’t show signs of chromosome damage during tests.
Moreover, other inherited bone marrow failure syndromes must also be considered. Some genetic disorders like dyskeratosis congenita, reticular dysgenesis, and Diamond-Blackfan anemia directly affect the bone marrow. They are usually associated with bone marrow failure, but they can be distinguished from Fanconi anemia by their specific symptoms and gene mutations.
Additionally, the doctor should also exclude De novo myelodysplastic syndrome, a condition where the bone marrow doesn’t produce enough healthy blood cells and could show similar symptoms to Fanconi anemia. Still, it doesn’t show susceptibility to chromosomal breakage test.
There could also be cases where a decrease in all types of blood cells is caused by exposure to harmful substances, like chemotherapy drugs, viral or bacterial infections, and chemicals. Unlike Fanconi anemia, the decrease in blood cells caused by these factors can be reversed when the harmful substance is removed.
Another condition to be considered is Paroxysmal nocturnal hemoglobinuria, a rare, acquired disorder that leads to the destruction of red blood cells. This condition can be distinguished from Fanconi anemia mainly because it isn’t inherited.
Finally, a few rare chromosomal breakage syndromes may also show symptoms that mimic Fanconi anemia. These could include Bloom syndrome, LIG4 syndrome, Ataxia-telangiectasia, Nijmegen breakage syndrome, Seckel syndrome, NHEJ1 deficiency, Warsaw breakage syndrome, and cohesinopathies. All these diseases also present abnormal chromosomal breakage tests. However, specifics in symptoms or genetic sequencing can help in differentiating these conditions from Fanconi anemia.
What to expect with Fanconi Anemia
Fanconi’s anemia is a serious health condition that often leads to poor health outcomes. The main cause of death in many children suffering from this condition is severe aplastic anemia, especially when it’s not diagnosed before 10 years of age. Besides, bacterial and fungal infections also often cause death in many children with Fanconi’s Anemia.
While advancements in medicine in developed countries have improved survival rates by reducing deaths from bleeding or infection complications, surviving into adulthood, unfortunately, increases the risk of developing cancer. Most people with Fanconi’s anemia eventually develop acute myelogenous leukemia or myelodysplasia.
The only known cure for these patients is allogeneic bone marrow transplant (BMT), but they need regular check-ups even after the transplant to monitor for any signs of relapse. In a small study, it was seen that about 16% of the patients experienced a relapse, and the transplant did not work, i.e., graft failure, in another 16% of them.
Furthermore, most of these patients also have other birth defects. These additional health issues can include developmental delays, kidney problems, and microcephaly (a condition where a baby’s head is significantly smaller than the usual).
Possible Complications When Diagnosed with Fanconi Anemia
Fanconi anemia is a medical condition that can lead to several major complications, including aplastic anemia, myelodysplastic syndrome (a disorder of the bone marrow), acute myeloid leukemia, and certain specific solid tumors. Patients with this condition have a mutation in a gene responsible for DNA repair and controlling cell growth. This defective gene can increase their sensitivity to chemotherapy drugs, radiation, and viruses, leading to the uncontrolled growth of cells.
The types of cancer commonly associated with Fanconi anemia include squamous cell carcinomas of the head, neck, and upper esophagus, as well as carcinomas of the vulva, anus, and cervix. Patients with this condition have a 50 times higher risk of developing these cancer types when compared to people without this disease. Myelodysplastic syndrome is the most common result, with a risk level that is 6000 times higher than in the general population. The second most common outcome is acute myelogenous leukemia, with a risk 700 times higher than the average.
In terms of age-related effects, around 90% of people with Fanconi anemia suffer from bone marrow failure by the time they reach 40. This can lead to a condition known as pancytopenia, which is a decrease in the amount of red and white blood cells, as well as platelets. This disease also presents a range of endocrine disorders, either due to the disease itself or linked to treatments.
Common Complications of Fanconi Anemia:
- Aplastic anemia
- Myelodysplastic syndrome (disorder of the bone marrow)
- Acute myeloid leukemia
- Specific solid tumors
- Squamous cell carcinomas of the head, neck, and upper esophagus
- Carcinomas of the vulva, anus, and cervix
- Bone marrow failure by age 40
- Pancytopenia (decrease in red and white blood cells and platelets)
- List of endocrine disorders
Additionally, short stature due to growth hormone deficiency, hypothyroidism in about 60% of patients, adrenal dysfunction, glucose intolerance, dyslipidemia, infertility due to hypogonadism, and liver conditions are also possible complications from androgen therapy.
Preventing Fanconi Anemia
Understanding Fanconi anemia is important, especially if a child in your family is diagnosed since this is a disease that can be passed down through families. Genetic counseling can help the family better comprehend the condition and learn how to manage it. This, in turn, improves communication between the family and doctors and makes following medical advice easier.
Having a child with Fanconi anemia means the entire family should be tested, particularly the siblings. This helps in either identifying if someone else also has the disease in early stages or even avoiding the birth of a child with genetic defects in the future. It’s important to know that not all close relatives who carry this gene will have the same risk of developing cancer as the child with the disease. However, families with a pattern of marrying within their culture or community might still face this risk.
Running special tests to check if a sibling could be a successful match for a hematopoietic stem cell transplant, which is a treatment for the disease, is a common practice. Parents should know about the disease, its symptoms, and treatment plans. It’s important that parents fully understand any potential treatments, their benefits, risks, and impact on health. After detailed counseling, families are then better equipped to decide on the most suitable treatment plan for their child.











