What is Hemophilia B?
Hemophilia B, sometimes called Christmas disease, is the second most common type of hemophilia. It is caused by a problem in the F9 gene, which leads to a lack of a blood clotting protein called factor IX. This genetic issue can be inherited from parents or it can appear out of the blue due to a random mutation.
Hemophilia B predominantly affects males, but females who carry the faulty gene may also sometimes experience heavier than normal bleeding. Females with the F9 mutation can have different levels of factor IX; those with at least 50% of the normal level usually don’t show any symptoms.
The disease was named after Stephen Christmas, who was the first person diagnosed with Hemophilia B in 1952. The disorder is also known as “the royal disease” because it was common in royal families across Spain, Germany, England, and Russia. The severity of symptoms can range widely, with males with severe Hemophilia B experiencing serious, spontaneous bleeding, sometimes from birth. On the other hand, those with milder forms typically bleed more after injuries or surgery, and symptoms might not show up until later in life.
This article covers the genetic causes of Hemophilia B, its symptoms, possible complications, and the importance of genetic testing. Such tests can help with diagnosing the disease, evaluating how severe it is, finding out if a female is a carrier, and making informed decisions related to care during pregnancy and childbirth.
What Causes Hemophilia B?
Hemophilia A, B, and C are conditions caused by not having enough clotting factors VIII, IX, and XI. These are proteins that help blood to clot, and having too little of them can lead to excessive bleeding. The main reason for this is usually genetics, although these conditions can sometimes appear unexpectedly.
The genes related to clotting factors VIII and IX are located on the X chromosome. Hemophilia A and B are inherited in a pattern referred to as X-linked recessive. This means it primarily affects males who can give the gene to their daughters but can’t give it their sons. However, females can still be carriers of the gene, with a 50% chance it could be passed to their children. Females, though, typically don’t show symptoms as they usually have one normal allele for the clotting factor. Still, some women may show symptoms if they get mutated alleles from both parents or due to a process called lyonization, where one X chromosome becomes inactive.
In very rare cases, a person can develop what’s known as acquired hemophilia B. This happens when the body’s immune system develops antibodies that attack its own clotting factors. This form of hemophilia often happens in people with an existing autoimmune disease, cancer, or infection, such as HIV or hepatitis B or C. The preferred terminology for this type of hemophilia is “acquired factor inhibitor” or “acquired factor deficiency.”
Risk Factors and Frequency for Hemophilia B
Hemophilia is a rare condition that can affect anyone, regardless of where in the world they live or their ethnicity. The rate of occurrence is 1 in 125,000. For a specific variant of the disease called Hemophilia B, the occurrence rate stands at 3.8 in 100,000 male newborns and 5 in 100,000 males at birth. In some communities, the frequency of hemophilia can be significantly higher due to marriages within the same family.
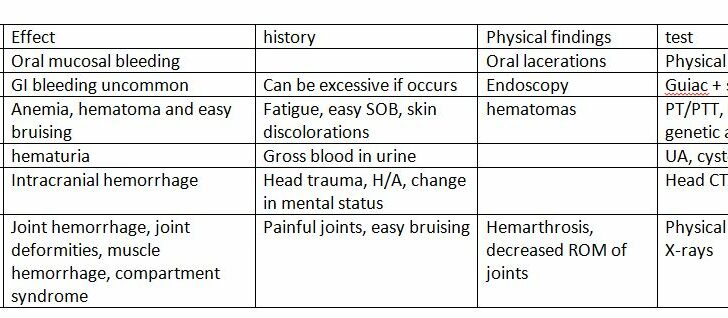
Signs and Symptoms of Hemophilia B
Hemophilia B is typically less severe than Hemophilia A. Its severity is dictated by the level of Factor IX in the blood, which plays a key role in blood clotting. Hemophilia B can be classified as severe, moderate, or mild.
Patients with severe hemophilia B typically have less than 1% of the normal amount of Factor IX in their blood. This can lead to spontaneous bleeding and symptoms may become noticeable in infancy or childhood with outbreaks following minor injuries. Symptoms can vary and include:
- Uncontrollable bleeding following circumcision, vaccinations, or mouth injuries
- Bleeding when starting to walk or crawl
- Bleeding in the brain, mouth, digestive tract, joints or muscles
Severe hemophiliacs also risk bleeding in organs like the liver, spleen, bladder, or kidneys. The most serious case is bleeding in the brain, which can cause seizures, muscle weakness, or poor feeding in newborns while older patients may experience headaches, vomiting, and lethargy.
Moderate hemophiliacs have a slightly higher amount of Factor IX in their blood, ranging from 1% to 5%. These patients typically do not bleed spontaneously but experience bleeding after trauma, injury, dental work or surgery. Recurrent joint bleeding can occur in up to 25% of cases.
Mild hemophilia B is noted when patients have between 5% and 40% of the normal amount of Factor IX. These patients only experience bleeding after significant trauma or surgery. Spontaneous bleeding is uncommon in these cases.
An important symptom of severe Hemophilia B is swelling and inflammation of joints (hemarthrosis) after bleeding. It accounts for around 80% of all hemophilia-related bleeds. The affected joints may feel warm and painful with limited movement. Repeated bleeding in the same joint can damage it permanently over time.
Frequent target areas for bleeding includes the major muscle groups. Bleeding in the muscles or bones can even lead to the formation of pseudotumors. Hemophiliacs may also experience nosebleeds or bleeding in the mouth and bowels. Bleeding in the bowel can look like appendicitis or other stomach disorders.
Testing for Hemophilia B
In about two-thirds of cases, those affected with hemophilia have a family history of the illness. If there’s a known family history of hemophilia or if a person has unexpected excessive bleeding after an injury, blood tests known as coagulation studies are necessary.
Medical professionals examine a person’s prothrombin time (PT), activated partial thromboplastin time (aPTT), and platelet levels when hemophilia is suspected. In Hemophilia B patients, aPTT is prolonged but PT and platelet levels are normal, suggesting an issue with the blood clotting process in the body. Since pregnancy and stress can falsely increase factor IX (a protein that helps blood clot) activity levels, these may need to be checked again after these factors are resolved.
It’s not uncommon for patients with minor factor deficiencies to display normal aPTT. Therefore, doctors may conduct a ‘mixing study’ with the patient’s blood to determine if a prolongation in aPTT is due to a lack of clotting factors or the presence of an inhibitor (an antibody against a clotting factor), disrupting normal functioning.
Men with a family history of hemophilia, patients with a clinical history suggesting hemophilia though they don’t have a known family history, and females who could be genetic carriers need to have factor activity tests. If the level of factor IX activity is below 40%, it establishes the diagnosis of Hemophilia B.
Genetic testing is also recommended for all hemophilia B patients as it might help predict those likely to develop inhibitors and identify carrier females in the family. Upon confirming a diagnosis of hemophilia B, routine lab check-ups should include screening for factor IX antibodies and infections related to blood transfusion, like Hepatitis and HIV.
Treatment Options for Hemophilia B
If a woman who carries hemophilia is pregnant with a boy, healthcare professionals need to be ready for the possibility of bleeding in both the mother and the baby. To prevent complications, doctors should avoid using certain instruments during delivery, like scalp electrodes, forceps, or a vacuum. When giving newborn screening tests, immunizations, and vitamin K, they should use the smallest needle possible, maintain pressure on the site after the procedure, and put ice on it for about 5 minutes. Circumcision should only take place after the diagnosis is confirmed and factor activity levels have been checked. Patients with hemophilia should receive their regular immunizations on time, and healthcare professionals should use the smallest needles possible, stick to one injection per limb, and apply pressure and ice for at least five minutes after giving the vaccine. If the patient receives supplemental factors, they should get the vaccine when their factor levels are high.
Regular exercise is very important for maintaining good health and preventing problems in patients with hemophilia. Healthcare professionals suggest non-contact sports like swimming, walking, golf, tennis, biking, archery, and table tennis. If patients are receiving supplemental factors and have the right safety equipment, they may be able to participate in higher-impact sports. However, this decision should be made with their healthcare team. Patients should avoid taking aspirin, non-steroidal anti-inflammatory drugs, and anticoagulants. Over-the-counter herbal remedies and supplements like fish oil can increase the risk of bleeding, so patients should discuss these with their healthcare provider. Patients with heart disease may need aspirin or an anticoagulant. A healthcare team will decide if these medications are necessary based on their risks and benefits. When traveling, patients should wear a medical alert bracelet, carry a supply of factor replacement, and know where the nearest hemophilia treatment center is.
Factor IX prophylaxis, a preventive treatment, aims to improve the quality of life by reducing episodes of hemarthrosis (joint bleeding), preventing the progression into hemophilic arthropathy (joint disease due to bleeding), and minimizing episodes of intracerebral (in the brain) and muscular bleeds. Patients with severe hemophilia and those with more than 2 bleeding episodes should receive prophylaxis. For those with mild-to-moderate hemophilia and no prior bleeding episodes, the need for prophylaxis is decided based on their physical activity level, and may only be needed intermittently.
The U.S. Food and Drug Administration (FDA) approved a new treatment in 2022 called Etranacogene dezaparovovec. This treatment contains a variant of the F9 gene which helps increase F9 activity. It provides a one-time treatment option for adults with hemophilia B who are on factor IX prophylaxis but still have severe bleeding incidents.
Management of acute bleeding in patients with hemophilia B primarily involves giving replacement factor IX. The amount given depends on the severity of the bleeding. The aim is to achieve a certain level of factor activity depending on the patient’s condition. The appropriate dose is calculated using a formula that takes into account the patient’s body weight, desired factor IX increase, and the factor accounting for the volume of redistribution.
Patients receiving factor IX replacement may develop a significant complication, the formation of IgG antibodies that block the activity of the replaced factor. These inhibitory antibodies develop as a response to foreign factors and they affect about 3% to 5% of patients with severe hemophilia B. They are much less common in patients with mild-to-moderate disease. Bleeding episodes can become more complicated due to the reduced response to factor infusions. Anaphylactic reactions may occur with factor IX inhibitors. In these cases, healthcare providers may consider alternatives such as plasmapheresis, bypassing products, or high-dose factor infusions.
When planning surgery for a patient with hemophilia, the operation, anesthesia, and hematology teams must collaborate closely with the lab and transfusion services. This pre-planning helps to properly administer factors, monitor levels, and determine the appropriate factor levels needed.
Future treatments such as gene therapy, cellular therapy, techniques to extend the half-life of factors, creation of factor alternatives, and applications of monoclonal antibodies are currently being developed to better manage hemophilia.
What else can Hemophilia B be?
There are several other conditions that can appear similar to hemophilia due to episodes of bleeding. These include:
- Deficiencies in certain blood clotting factors, for example, hemophilia A (factor VIII deficiency) and C (factor XI deficiency). Doctors can tell these apart by studying clotting factors in the lab and through genetic testing. Hemophilia A and B are inherited from the mother, while hemophilia C can be passed down from either parent.
- Deficiency of von Willebrand Factor (vWF), which is important for blood clotting. This condition leads to an issue with the formation of an initial clot, known as a platelet plug. People with this condition have a longer than normal bleeding time, normal or longer time for blood to clot, and a normal platelet (a type of blood cell that helps blood clot) count.
- Conditions that cause problems with the quantity or quality of platelets, which play a central role in blood clotting. These can cause bleeding in the skin or mucus membranes. Examples include immune thrombocytopenia, thrombotic thrombocytopenia, and hemolytic uremic syndrome. Usual clues for these conditions are an increased bleeding time and a decrease in platelet count.
- Disseminated intravascular coagulation, which causes both clot formation and bleeding. This condition can be triggered by various events such as severe infections, trauma, complications during childbirth, acute pancreatitis, certain types of leukemia, or a blood transfusion.
- Vitamin K deficiency, which can occur in newborns or in individuals on long-term antibiotics. This condition can actually cause an increase in the time taken for blood to clot as well as decrease in certain clotting factors and proteins, while keeping the platelet count normal.
- Scurvy, a lack of vitamin C, often causes swollen gums, unusual bleeding, joint pain, and slow wound healing.
- Ehlers-Danlos syndrome, a genetic disorder that affects the body’s ability to produce collagen, a protein that strengthens skin. This often causes bleeding from mucous membranes, super-stretchy skin, and extremely flexible joints.
Healthcare professionals should be vigilant and look for signs of inconsistencies in accounts of injury when identifying possible cases of child abuse, which can sometimes be misidentified as hemophilia. Significantly, child abuse might also include signs of varying healing stages in wounds, malnutrition, brain bleeding, bleeding in the eyes, and signs of sexual abuse like sexually transmitted infections and urinary tract infections.
What to expect with Hemophilia B
Judith Graham Pool, an American doctor and 1964 Nobel Prize winner, made a major breakthrough by extracting a substance called cryoprecipitate from plasma that contained more clotting factors. This advancement greatly improved the outlook for people with hemophilia. Before 1970, someone with hemophilia typically lived only to their early teens. Then, in 1970, researchers managed to extract the first clotting factor from plasma, a development which vastly improved the quality of life and survival rates for people with hemophilia. Before these advances, the only treatment available involved transfusions of whole blood or plasma.
However, problems emerged in the early 1980s. Many patients receiving this new treatment contracted HIV and hepatitis C. By 1992, these diseases affected a staggering 80% to 85% of hemophilia patients who had been treated with clotting factors. Fortunately, better screening techniques were developed which made plasma-derived products safer and reduced the risk of transmission.
Today, the outlook for patients in developed countries is much brighter. Patients can expect to live nearly as long as anyone else if they can get early access to clotting factor replacements and preventative treatment starting at ages 1 to 2 for severe cases. On the other hand, the lack of health systems and resources in developing countries means that hemophilia patients still die at double the rate of healthy men. And stark differences in care still exist: in mid-to-high income countries, someone born with hemophilia has a 64% lower chance of living a normal lifespan. That gap jumps to 77% in middle-income countries, and up to 93% in low-income areas.
Possible Complications When Diagnosed with Hemophilia B
The primary complications of hemophilia, a blood clotting disorder, include recurrent bleeding into joints, bleeding into the brain, HIV and Hepatitis C infections, and the body developing resistance to clotting factor treatment. Continuous bleeding into the joints can cause inflammation and enlargement of the joint lining, leading eventually to severe joint damage. Bleeding into the brain can result in long-term neurological disabilities.
HIV and Hepatitis C infections can lead to serious conditions, even death and liver cancer. Some people might also have a low blood volume shock due to bleeding into the muscles in the lower back, or difficulty breathing due to bleeding in the throat. People with hemophilia tend to have higher rates of mental, social, and physical struggles, along with depression, compared to healthy individuals. Also, children with hemophilia may experience developmental delays.
Most Common Complications:
- Recurrent bleeding into joints
- Bleeding into the brain
- HIV and Hepatitis C infections
- Body developing resistance to clotting factor treatment
- Low blood volume shock due to muscle bleeding
- Breathing difficulty due to throat bleeding
- Higher rates of mental, social, and physical struggles
- Depression
- Developmental delays in children
Preventing Hemophilia B
Hemophilia is a medical condition that prevents blood from clotting properly, which can sometimes lead to serious issues. People with hemophilia have certain components missing from their blood which are necessary for clotting. Hemophilia A patients lack something called factor VIII, and hemophilia B patients lack factor IX. This disease often causes easy bruising, abnormal bleeding after dental or surgical procedures, or bleeding into a joint.
While there isn’t a cure for hemophilia, treatments are available. One common treatment is scheduled replacement of the missing factor in the blood. Some people only get this treatment when they suffer an injury or need surgery. If hemophilia patients have an injury, they can manage their immediate pain by using cold packs, resting, wearing splints, and taking certain pain relief medications like acetaminophen and codeine. However, certain medications like anticoagulants, aspirin, and some types of anti-inflammatory drugs can cause problems with blood clotting.
People with hemophilia should stay current on their vaccinations, but healthcare providers should take extra precautions when giving them injections. This includes using a small needle, applying ice packs, and applying pressure for 5 minutes after the injection. It is also recommended to give only one injection per limb. Patients who are receiving treatment to replace missing blood factors should get their vaccines when these factors are at their highest levels in their blood.
Good oral hygiene is also important for hemophilia patients. They should brush and floss at least twice a day. Regular physical activity is also beneficial, especially non-contact sports. If a patient wishes to participate in contact sports, they should first discuss this with their healthcare team.
For pregnant women carrying a baby diagnosed with hemophilia, there is no evidence suggesting either C-section or vaginal delivery is preferable. However, healthcare providers should avoid using forceps or vacuum extraction during birth, as these methods can increase the risk of brain bleeding and a blood collection under the scalp. Delaying circumcision until after the diagnosis is confirmed or ruled out is also advisable.











